NoverIsolation and structure elucidation of most of the major cannabinoid constituents—including Δ 9-tetrahydrocannabinol (Δ 9-THC), which is the principal psychoactive molecule in Cannabis sativa—was achieved in the 1960s and 1970s. It was followed by the identification of two cannabinoid receptors in the 1980s and the early 1990s and by the identification of the endocannabinoids shortly thereafter. There have since been considerable advances in our understanding of the endocannabinoid system and its function in the brain, which reveal potential therapeutic targets for a wide range of brain disorders.
Raphael Mechoulam, Lumír O. Hanuš, Roger Pertwee and Allyn C. Howlet
The plant Cannabis sativa and its many preparations (for example, marijuana, hashish, bhang and ganja) have been used for millennia for recreation (and at times for the achievement of religious ecstasy) as well as in medicine. In ancient China, cannabis was prescribed (together with other plants, as is customary in Chinese medicine) for numerous diseases, but it was noted that when taken in excess it could lead to ‘seeing devils’. In Assyria (about 800 bc), it was named both gan‑zi‑gun‑nu (‘the drug that takes away the mind’) and azallu (when used as a therapeutic). In India, ancient Persia and medieval Arab societies, cannabis use proceeded along these two divergent routes1. In many countries, hemp — a strain of Cannabis sativa that does not cause psychoactivity — was grown for its durable fibres. Our present-day society follows a long tradition of recreational, industrial and medical cannabis use.
Cannabinoid discovery — early history
The behavioural effects of cannabis, in several animal species as well as in humans, were observed in the mid-nineteenth century2 (FIG. 1). These experimental observations led to the first attempts to isolate the active constituents of the plant, as had already been done with other plants that had known neuropharmacological activity — for example, the isolation of morphine. A prize was even awarded in 1855 for the ‘successful’ accomplishment of this project. However, the first isolation of a plant cannabinoid — named cannabinol (CBN) — was not achieved until the end of the nineteenth century. Its structure was elucidated much later, in the 1930s, by the groups of Cahn and Todd in the United Kingdom and by Adams in the United States, when a further component, cannabidiol (CBD), was isolated; however, its structure could not be elucidated at that time. Although considerable effort was invested on the isolation and the elucidation of the structure of the main psychoactive constituents of cannabis, this goal was not reached at that time. A synthetic compound, Δ6a,10a‑tetrahydrocannabinol (Δ6a,10a-THC), showed pharmacological activity that paralleled the activity of cannabis extracts. Therefore, it was assumed that Δ6a,10a-THC was chemically related to the active compounds of the plant (FIG. 2). Much of the early research in this area was done using synthetic Δ6a,10a-THC, which is now known to be considerably less potent than the actual natural product. The chemical and pharmacological work that was carried out until the mid 1940s has been reviewed elsewhere3–5. Some Δ6a,10a-THC analogues were even tested in humans. In light of recent media reports about the action of cannabinoids in paediatric epilepsy, it is of interest to note that a derivative of synthetic Δ6a,10a -THC (at doses of 1.2–1.8 mg daily) was administered to a small number of children with epilepsy and showed positive results. Historical cannabis use in medicine over the ages and early chemical investigations are reviewed in REF. 1.
The reasons for the lack of progress were mostly technical. We now know that cannabinoids are present in cannabis as a mixture of many closely related constituents — over 100 — which were difficult to separate using the methods that were available in the nineteenth and early twentieth centuries. As the active constituents of cannabis were not available in pure form, there was very little biological or clinical work done in this area from the late 1940s until the mid 1960s.
By the 1960s, chromatography methods were well developed for the isolation of pure compounds from mixtures and the availability of novel spectrometric methods meant that the elucidation of the structure of these compounds was possible. Indeed, many cannabinoids were isolated, including Δ9-THC, which was reported by Gaoni and Mechoulam in 1964 (REF. 6) (FIG. 2). Their structures were mainly elucidated using NMR, which was a modern method at the time. Several total syntheses of these compounds have been reported and most cannabinoids are now available as both natural and synthetic products. The chemical work until the mid 1970s is reviewed in REF. 7.
The next step in cannabinoid research was the elucidation of the metabolism of Δ9-THC and later of CBD. The major metabolic pathway of Δ9-THC is hydroxylation, which leads to the formation of an active metabolite, followed by its further oxidation to an inactive acid, which then binds to a sugar molecule. These acid-derived metabolites are stored in fatty tissues and are slowly released8. Indeed, the major final Δ9-THC metabolite (a carboxylic acid that is present as a glucuronide) can be detected in human urine for several weeks after cannabis use (FIG. 2).
Early neuropharmacology
The advances in the chemistry of plant and synthetic cannabinoids led to renewed interest in their neuropharmacology. Loewe5 had found that cannabis extracts (presumably containing high levels of what is now known to be Δ9-THC and additional phytocannabinoids) can induce catalepsy in mice and that CBN can also produce this effect, albeit much less potently than the impure THC isolated from the resin. It was these findings that prompted the development by Pertwee9 in 1972 of a quantitative in vivo assay for psychotropic cannabinoids, known as the ring test, in which the proportion of time that a mouse placed across an elevated horizontal ring remains immobile or cataleptic is measured over a 5 minute period9. Martin10 later used this assay, along with three other bioassays, in what came to be known as the ‘mouse tetrad assay’10. These other assays provide measures of cannabinoid-induced hypokinesia, hypothermia and antinociception in mice, using a tail flick or hot plate test. The mouse tetrad assay is a useful in vivo screen for psychotropic cannabinoids, all of which, in contrast to many other types of drugs, generally show similar potency in all four of these bioassays. It was also discovered in the 1940s that cannabinoids can elicit central excitant activity in rabbits and mice and corneal arreflexia in rabbits, and that some phytocannabinoids, particularly CBD, can prolong barbiturate-induced sleep by a mechanism that was subsequently discovered to involve the inhibition of certain cytochrome P450 (CYP) enzymes10.
Following its identification as the main psychoactive constituent of cannabis, Δ9-THC attracted particular attention10,11; for example, results obtained from several investigations on humans indicated that when Δ9-THC was taken orally or intravenously or when it was inhaled in smoke, it showed substantial potency at producing psychological changes similar to those reportedly experienced in response to recreationally consumed cannabis11. A few other phytocannabinoids, such as CBN, were found to induce cannabis-like effects in humans with low potency (an exception being Δ8-THC but there is usually very little Δ8-THC in cannabis)11.
It is noteworthy that one synthetic analogue of Δ9-THC, nabilone (Cesamet; Valeant Pharmaceuticals North America) was approved in 1981 as a medicine for the suppression of the nausea and vomiting that is produced by chemotherapy12. Synthetic Δ9-THC, dronabinol (Marinol; Solvay Pharmaceuticals, Inc) subsequently entered
the clinic as a licensed medicine, in 1985 as an antiemetic and in 1992 as an appetite stimulant12. Claims from patients that cannabis can ameliorate unwanted symptoms of multiple sclerosis also encouraged the development of the cannabis-based medicine naviximols12 (Sativex; GWPharma), which contains both Δ9-THC and the non-psychoactive CBD; this was first licensed as a medicine in 2005 in Canada for the relief of pain experienced by adult patients suffering from multiple sclerosis or advanced cancer, and subsequently as a medicine to ameliorate spasticity caused by multiple sclerosis12.
Discovery of the cannabinoid receptors
Although a considerable amount of pharmacological work was done on the activity of Δ9-THC, its mechanism of action was not elucidated for more than 20 years after its identification. Indeed, it was originally thought that the mode of action of Δ9-THC was nonspecific in nature and that it might involve interactions with lipid membranes. However, although the stereospecificity of the action of Δ9-THC and related synthetic cannabinoids13,14, as well as pharmacological studies, in humans and animals had suggested a putative cannabinoid receptor15,16, it was not until the 1980s that evidence for a protein receptor was sought.
As the family of known G proteins expanded in the late 1970s and early 1980s, so did the list of receptors for hormones and neurotransmitters to which they could couple. Agonists of opioid, muscarinic, cholinergic and α‑adrenergic receptors resulted in inhibition of Gs‑stimulated adenylyl cyclase17–19, and functional homology with these neuromodulators led to the discovery that cannabinoids also inhibited this enzyme20–22 by a pertussis toxin-sensitive mechanism23. This clearly indicated that the cannabinoid receptor was a G protein-coupled receptor (GPCR).
From the structure–activity relationship (SAR) established using cannabimimetic compounds from Pfizer Central Research, the Howlett laboratory identified CP55940 (FIG. 2) as a highly potent cannabinoid analogue and, in 1988, reported the determination and characterization of a cannabinoid receptor from the brain for which the criteria for a high-affinity, stereoselective receptor in brain tissue had been fulfilled24. Competitive displacement of [3H]CP55940 from its target in rat brain membranes by cannabinoid agonists was enantioselective and followed the order of potency for both Gi‑mediated inhibition of adenylyl cyclase as well as antinociception in several rodent models24–27. Later, signal transduction assays were used to ultimately deorphanize a 7‑transmembrane receptor now known to be the cannabinoid receptor 1 (CB1; also known as CNR1)28,29.
Discovery of endocannabinoids and CB2
Receptors are mostly activated by endogenous molecules, and therefore, there was a strong reason to look for endogenous cannabinoids. As Δ9-THC and its related compounds that bind to the CB1 are lipids, it was reasonable to assume that any endogenous cannabinoids would also be lipids. In order to isolate putative endogenous cannabinoid compounds, the ability of porcine brain extracts to displace a novel, highly potent radiolabelled cannabinoid probe, [3H]HU‑243, bound to CB1 was tested in the Mechoulam laboratory. The fractions that inhibited the binding of [3H]HU‑243 to the cannabinoid receptor were purified by a series of chromatographies, which ultimately led to the generation of a minute amount of a single compound, an amide of arachidonic acid — arachidonoyl ethanolamide — which was named anandamide; this was the first endocannabinoid to be identified30. The structure of anandamide (FIG. 3) was established by mass spectrometry, NMR spectroscopy and by its synthesis30. Anandamide was found to have inhibitory activity that was equivalent to that of Δ9-THC and was subsequently shown to have cannabimimetic activity as it inhibited the twitch response of isolated mouse vasa deferentia30.
In the meantime, a second receptor, CB2 (also known as CNR2), had been identified by sequence homology31 and was presumed to be mainly present in the periphery; therefore, a search for a ‘peripheral’ endogenous agonist was initiated. Using the same techniques that were used to isolate anandamide, it was possible to isolate an ester of arachidonic acid — 2‑arachidonoyl glycerol (2‑AG)32 — from canine intestines (FIG. 1). This compound was unexpectedly found to bind CB1 and CB2 and to inhibit adenylyl cyclase with a potency similar to that of Δ9-THC. 2‑AG also shared the ability of Δ9-THC and anandamide to inhibit electrically evoked contractions of isolated mouse vasa deferentia; however, it was less potent than Δ9-THC32. Following administration
to mice, both anandamide and 2‑AG caused the typical tetrad of effects produced by Δ9-THC: antinociception, immobility, reduction of spontaneous activity and lowering of rectal temperature.Although a few additional endocannabinoids have been reported, none of them has been confirmed as a natural endocannabinoid.
Anandamide is a partial agonist for CB1 and CB2 and shows less relative intrinsic activity (also known as relative intrinsic efficacy) and affinity for CB2 than for CB1. 2‑AG shows greater potency and efficacy than anandamide as a CB1 agonist and greater potency than anandamide as a CB2 agonist33. In addition, it has been found that both endocannabinoids interact with certain non-CB1 and non-CB2 receptors and ion channels33. In the past few years, lipoxin A4 and a new family of peptides (known as pepcans) have been reported to target CB1 as allosteric modulators34,35 and the peptide hemopressin, which is a putative brain constituent, has been found to lower pain via action on a cannabinoid receptor36.
Synthesis of cannabinoid analogues that have high affinity and specificity for CB2 was achieved in the mid to late 1990s37,38 and led to the discovery of the role of CB2 in immunosuppression, neuroprotection and neuropathic and inflammatory pain. This consequently led to considerable interest in developing and investigating CB2-selective agonists39–43.
Both anandamide and 2‑AG are synthesized on demand, often in response to increased concentration of intracellular calcium44, and it is now generally accepted that one important role of these endocannabinoids, although possibly only of 2‑AG, is to function as retrograde synaptic messengers that can prevent the development of excessive neuronal activity in the central nervous system and thereby contribute to the maintenance of homeostasis in both health and disease45. Thus, there is good evidence that neurotransmitters, such as glutamate, produce postsynaptic increases in the concentration of intracellular calcium in a manner that can induce postsynaptic biosynthesis and release of anandamide or 2-AG into the synapse. In turn, this induces subsequent endocannabinoid-induced activation of presynaptic CB1, which causes an inhibition of the neuronal release of glutamate, γ‑aminobutyric acid or other neurotransmitters in brain areas that include the cerebral cortex, hippocampus, ventral tegmental area, substantia nigra, hypothalamus and cerebellum46–48. There is also evidence that, when produced postsynaptically in response to the activation of postsynaptic metabotropic glutamate receptor 5 (MGLUR5) , anandamide activates postsynaptic transient receptor potential cation channel subfamily V member 1 (TRPV1) channels48. It is also noteworthy that results obtained from in vivo experiments with rats suggest that retrograde 2‑AG signalling that is triggered by the activation of MGLUR5 can suppress pain sensitivity49. The endocannabinoid retrograde transport mechanism and modulation of synaptic transmission have not yet been fully elucidated46–48.
Search for antagonist ligands
The holy grail for cannabinoid synthetic chemists was an antagonist that could block the effects of Δ9-THC. It seems quite unusual that no natural product or structurally related analogue emerged to block the cannabinoid receptors. Before the advent of gene knockout techniques, it was difficult to establish whether a pharmacological effect was mediated by a receptor if a selective antagonist for that receptor had not been developed. Thus, one can imagine the excitement generated at an International Cannabinoid Research Society meeting in 1993 when a team of researchers from the French pharmaceutical company Sanofi Recherche announced their discovery of an antagonist for CB1, SR141716A50. This compound was radiolabelled to investigate receptor pharmacology51 and was soon modified to develop the first ligands for in vivo imaging52. The discovery of an antagonist (SR141716A), which was in fact subsequently identified as an inverse agonist, helped to characterize additional cellular signalling pathways for CB1 (REFS(50),(51),(53)–(55)). More importantly, an antagonist could finally be used to identify animal behaviours that were truly due to CB1 activation56–58. Indeed, the syndrome of ‘dependence’ on cannabinoid agonists was first shown in an animal model after precipitated withdrawal using SR141716A59,60. Within a short period of time, industrial laboratories and academic research groups reported the synthesis of additional CB1 antagonists and inverse agonists61–64.
The first CB2-selective antagonists AM630 (also known as iodopravadoline) and SR144528 emerged in the mid 1990s65,66 and increased the ability to discern novel actions that could be attributed to CB2, including actions observed in liver Kupfer cells67, microglial cells and astrocytes68,69 and in the gastrointestinal system70, among others. Since that time, there has been considerable progress towards the development of highly selective and potent CB2 antagonists41,71.
SR141716A (also known as rimonabant) is used therapeutically for the treatment of obesity-related metabolic syndrome components, including dyslipidaemia and diabetes72–74. SR141716A was marketed in Europe but failed to gain approval from the US Food and Drug Administration. As might be predicted, a drug that blocks CB1 neuromodulation at synapses for the major stimulatory (in the case of glutamate) and inhibitory (in the case of GABA) transmitters throughout the brain would be likely to produce multiple ‘off-target’ effects. One such side effect, which was reported in 2009, was an increase in reported signs of depression in vulnerable individuals treated with SR141716A75,76. It could be argued that the benefit to risk ratio in a morbidly obese patient population might mitigate the concerns about depression. However, the drug was withdrawn from the market and similar analogues from other pharmaceutical companies were taken out of the development pipeline. Nevertheless, the development of SR141716A by Sanofi– Aventis can be considered to be a major contributor to our understanding that CB1 is present and functional in tissues such as adipose, liver and pancreas under pathological conditions of high-fat diet or obesity77. This new understanding of the role of CB1 in metabolic regulation has inspired the search for novel antagonists that fail to gain access to the brain78,79. An alternative clinical strategy would be to screen for individuals who might be most susceptible to the limbic effects of CB1 antagonists before selecting a treatment modality80.
Endocannabinoid neuropharmacology
The discovery that anandamide and 2‑AG are endocannabinoids prompted research to identify the biochemical processes that are responsible for both their biosynthesis and their metabolism. This research showed that these two endocannabinoids are synthesized ‘on demand’ rather than stored, and it identified biosynthetic and metabolic pathways for both of them81–83. Thus, it has been discovered that 2‑AG is formed from diacylglycerol (DAG) in a process that is catalysed by sn1‑specific DAG lipase‑α and lipase-β, and that the main biosynthetic pathway for anandamide involves the formation of N‑arachidonoyl phosphatidylethanolamine (NAPE) from phosphatidylethanolamine and phosphatidylcholine, which is catalysed by an as yet uncharacterized calcium-dependent transacylase enzyme. This is then followed by the conversion of NAPE to anandamide in a single step that is catalysed by NAPE-selective phospholipase D and/ or in two or three steps that are catalysed by other enzymes. It has also been found that, following their release, anandamide and 2‑AG are mainly metabolized to arachidonic acid, the major metabolizing enzymes being fatty acid amide hydrolase (FAAH) for anandamide and monoacylglycerol lipase (MAGL) for 2‑AG81,82. Other endocannabinoid-metabolizing enzymes include FAAH‑2 for anandamide, α,β‑hydrolase domain-containing 6 (ABDH6) and ABDH12 for 2‑AG, and cytochrome P450 enzymes, lipoxygenases and cyclooxygenase 2 for both of these endocannabinoids81,82. The physiological relevance of the lipoxygenase and cyclooxygenase derivatives of anandamide and 2‑AG is not yet clear. It is also noteworthy that anandamide and 2‑AG can undergo cellular uptake following their release, although whether this process is mediated by a transporter is currently unclear81,82.
It is now recognized that, although engineering exogenous cannabinoids provided insights into receptor usage and linked functional events, the intracellular and extracellular actions and fate of endocannabinoids versus those of exogenously introduced cannabinoids may differ and have different physiological consequences 33,44. It is also recognized that many cannabinoid receptor ligands also interact with a wide range of non-cannabinoid receptor targets and that, irrespective of whether they are endogenous, synthetic or plant cannabinoids, the pharmacological profiles of these compounds often vary considerably from each other33,44.
The endocannabinoid receptors, the endocannabinoids and their biosynthetic and biodegrading enzymes constitute what has come to be known as the endocannabinoid system, the discovery of which prompted a search for its physiological and pathophysiological roles. This search revealed that there are several disorders in which endocannabinoids are released to their receptors in an ‘autoprotective’ manner that ameliorates unwanted effects of these disorders82–84. It also raised the possibility that increasing extracellular levels of a released endocannabinoid by inhibiting metabolizing enzymes such as FAAH or MAGL, or by inhibiting the cellular uptake of anandamide, might prove to be an effective therapeutic strategy to manage some of these disorders, which include multiple sclerosis, Parkinson’s disease, schizophrenia, hypertension, inflammatory bowel diseases, pruritus, Alzheimer’s disease, depression, obsessive compulsive disorder and cancer82–84.
The discovery of the endocannabinoid system also led to a reinvestigation of the interactions of plant and synthetic cannabinoids with this system and other biochemical entities. As a result, evidence has emerged that Δ9-THC targets receptors other than CB1 (REFS(85)–(87)). For example, at submicromolar concentrations, Δ9-THC has also been found to have several effects: first, it has been found to activate CB2, albeit with less efficacy than it activates CB1 (REF.(88)); second, it has been found to activate the deorphanized GPCRs GPR18 (REF.(89)) and GPR55 (REF.(33)), the cation channels TRPA1 and TRPV2 (REFS(90)) and the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ)33; third, it has been found to block the activation both of 5-hydroxytryptamine 3 (5HT3) ligand-gated ion channels33,85 and of TRPM8 cation channels90; and, last, it has been found to enhance the activation both of α1 subunits and α1β1 dimers of human glycine ligand-gated ion channels and of native glycine receptors in rat isolated ventral tegmental area neurons33. There have also been reports that submicromolar concentrations of Δ9-THC can inhibit the enzyme lysophosphatidylcholine acyl transferase11, that it can increase the activity
of phospholipase C, which can catalyse the production of DAG and phospholipase A2 (REF.(11)) and that it can both inhibit the uptake of adenosine by cultured microglia and macrophages and affect the synaptosomal uptake of 5‑hydroxytryptamine (it inhibits this process), of noradrenaline (it enhances this process) and of dopamine (it both enhances and inhibits this process)85,87. In addition, at higher concentrations, Δ9-THC has been found to affect several other such pharmacological targets85,87. For example, at concentrations between 1 μM and 10 μM, it has been reported to enhance the activation of β‑adrenoceptors, to function as a negative allosteric modulator of μ-and δ‑opioid receptors, to activate the cation channels TRPV3 and TRPV4 and to inhibit T‑type calcium (Cav3) and potassium (Kv1.2) voltage-gated ion channels, as well as conductance in Na+ voltage-gated ion channels. In this concentration range, Δ9-THC has also been reported to inhibit the enzymes lipoxygenase, Na+–K+-ATPase and monoamine oxidase, as well as the cytochrome P450 enzymes CYP1A1, CYP1A2, CYP2B6 and CYP2C9, to inhibit noradrenaline-induced melatonin biosynthesis, and to activate or to inhibit Mg2+-ATPase85,87.
Perspectives
There has been much progress in our understanding of the plant cannabinoids and of CB1 and CB2. We have identified endogenous lipid mediators that act on these receptors to regulate multiple pathways of cellular signalling. We have discovered synthetic agonists and antagonists for these receptors as well as allosteric modulators of CB1. However, there is still much more knowledge to be gained and challenges to be met in the fields of cannabinoid receptor neuroscience, pharmacology, molecular biology and cannabinoid medicine.
We now need to understand how the endocannabinoid receptors interact with other proteins in complexes that regulate differentiated functions both at the cell surface and in intracellular organelles, particularly in the brain91–93.
Dozens of endogenous molecules, with structures that resemble those of the endocannabinoids, have been discovered in the brain94,95. The activity of most of these molecules is not known. Some of those that have been investigated show activities that have therapeutic potential; for example, arachidonoyl serine is a vasodilator96 and is neuroprotective after brain injury as it reduces apoptosis97. It leads to proliferation of neural progenitor cells in vitro and maintains these cells in an undifferentiated state in vitro and in vivo. Although it does not bind to CB1 and CB2, its activity is blocked by CB2 antagonists98. This raises questions, such as what is the relationship of such endocannabinoid-like compounds to the endocannabinoid system and what are the physiological roles of these molecules in the brain?
Pucci et al.99 have investigated the possible epigenetic regulation of skin differentiation genes by phytocannabinoids99. CBD was found to increase DNA methylation of the keratin 10 gene. Remarkably, CBD also reduced keratin 10 mRNA levels by a CB1-dependent mechanism. Thus, in this system, CBD is apparently a transcriptional repressor that can control cell proliferation and differentiation. As anandamide has also been found to have epigenetic properties100, it is of interest to determine the extent, if any, of transcriptional control by endocannabinoids by epigenetic mechanisms.
Although various methods have been used to enhance endocannabinoid levels in vivo (even in patients)82,101, neither anandamide nor 2‑AG have been administered to humans. In addition, only a small number of clinical studies have been carried out using plant cannabinoids. A notable exception is the recent successful clinical trial using CBD in schizophrenic patients101. Although it is widely mentioned in the general media that cannabis with a high concentration of CBD is therapeutic in paediatric epilepsy and that ‘medical marijuana’ is indeed of value in such cases102, there have not been any recent clinical trials reported, although several such trials are ongoing (an anti-epileptic trial of CBD in adults was reported 34 years ago103).
In a recent review, Pacher and Kunos84 suggested that “modulating endocannabinoid system activity may have therapeutic potential in almost all diseases affecting humans”. They supported this strong statement with a long list of examples, although these examples were mostly obtained in vitro or from in vivo experiments in animals84. If this summary of effects is shown to reflect actions in human patients, is the endocannabinoid system going to bring a revolution in therapy? This might be the case as investigators are now able to target multiple cell-specific synthetic and biotransformation enzyme pathways that can adjust the levels of endocannabinoid ligands with some degree of tissue selectivity. In addition, aside from the agonist and antagonist ligands for cannabinoid receptors, researchers can now target cell type-specific allosteric modulators and receptor-associated proteins. Thus, there is great promise for the future of cannabinoid research.
Raphael Mechoulam is at the Institute for Drug Research, Medical Faculty, Hebrew University, Jerusalem, 91120, Israel.
Lumír O. Hanuš is at the Institute for Drug Research, Medical Faculty, Hebrew University, Jerusalem, 91120, Israel.
Roger Pertwee is at the Institute of Medical Sciences, University of Aberdeen, Aberdeen AB25 2ZD, Scotland, UK.
Allyn C. Howlett is at the Department of Physiology and Pharmacology, Wake Forest University Health Sciences, One Medical Center Blvd, Winston-Salem, North Carolina 27157, USA.
Acknowledgements
Research in the laboratory of R.M. was supported by the Kessler Family Foundation, Boston, USA, and by a grant from US National Institute on Drug Abuse (NIDA), DA‑9789. The research of R.P. was supported by NIDA grants DA‑3934, DA‑9789 and DA‑3672 and GW Pharmaceuticals and the research of A.H. was supported by NIDA grant DA-3690.
Competing interests statement The authors declare no competing interests.
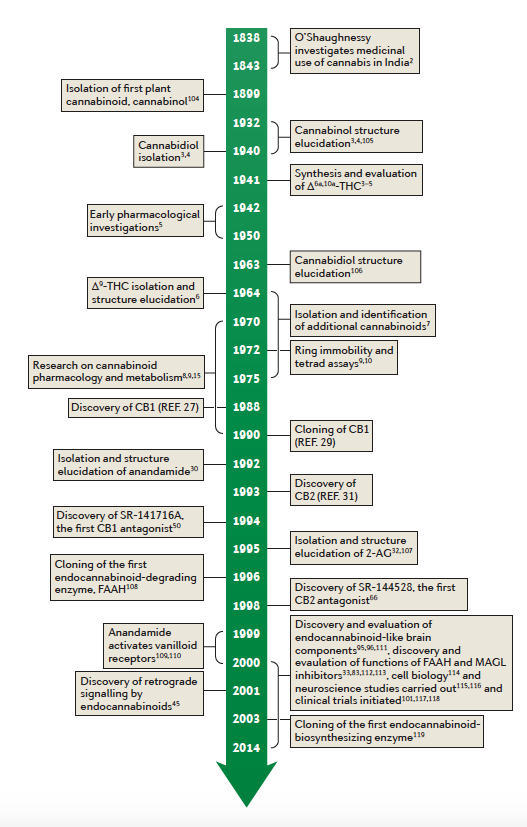
View Larger
Figure 1 | Cannabinoid and endocannabinoid research — a timeline. Almost all early research
was devoted to clarification of cannabinoid chemistry3,4,104,105, and pharmacology was mainly done using synthetic compounds5. Following the isolation and structure elucidation of the plant cannabinoids, particularly of cannabidiol106 and of Δ9-tetrahydrocannabinol (Δ9-THC)6, pharmacological and physiological work was initiated8,9,15. The identification of cannabinoid receptors24,29,31, of endogenous cannabinoids30,32,107 and of receptor antagonists50,66 made possible extensive pharmacological
and neurobiological research leading to cloning of the anandamide-degrading
enzyme fatty acid amide hydrolase (FAAH)108, the discovery of retrograde signaling by
2-arachidonoyl
glycerol (2‑AG)45, the discovery of allosteric sites on cannabinoid
receptor 1
(CB1)33, the discovery that endocannabinoids bind to receptors other than CB1 and CB2
(REFS 109–111), the discovery and evaluation of endocannabinoid-like molecules in the brain95,96
and the discovery and function of inhibitors of the endocannabinoid-degrading enzymes112,113.
Cell biology114 and neuroscience115,116 investigations were also carried out, and clinical trials were
initiated101,117,118. Cloning of DAG lipase was also reported119.
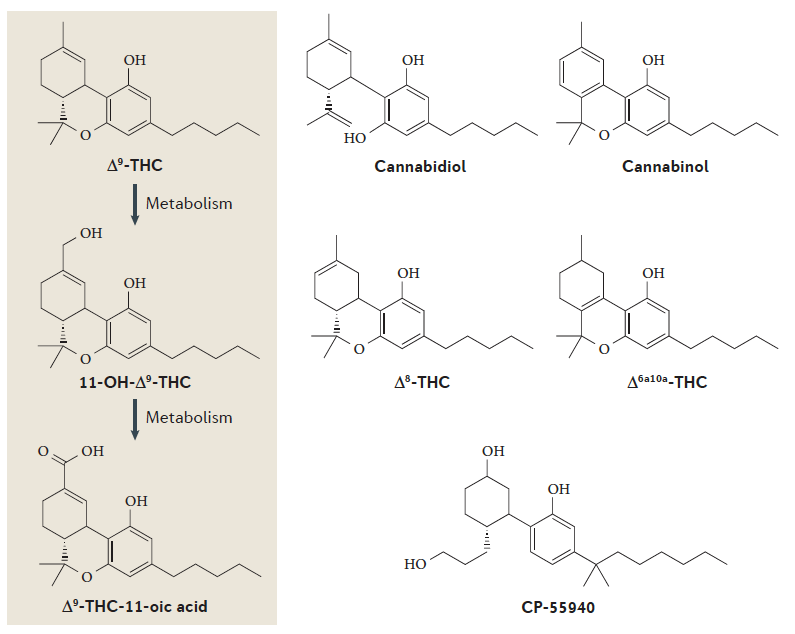
View Larger
Figure 2 | A major metabolic pathway of Δ9-THC and the structures of some plant and synthetic cannabinoids. a | The major psychoactive cannabis constituent, Δ9-tetrahydrocannabinol (Δ9-THC), is first metabolized by enzymatic hydroxylation to produce psychoactive 11‑hydroxy- Δ9-THC (11-OH-Δ9-THC) and then by enzymatic oxidation to non-psychoactive Δ9-THC-11‑oic
acid, which is stored in fatty tissues as a glucuronide and is slowly released. The glucuronide may
be detected in the urine for several weeks after a single cannabis use. b | The structures of some plant
and synthetic cannabinoids. Δ9-THC, the plant constituents cannabinol and Δ8-THC, and synthetic
Δ6a,10a-THC and CP‑55940 cause cannabis-type psychoactivity, wherease cannabidiol does not.
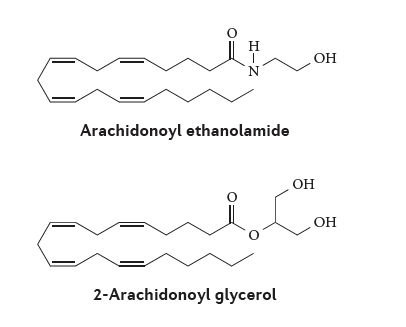
View Larger
Figure 3 | Structures of the main endocannabinoids, anandamide and 2‑AG, which bind to
Figure 3 | Structures of the main endocannabinoids, anandamide and 2‑AG, which bind to
CB1 and CB2 endocannabinoid receptors. Arachidonoyl ethanolamide (also known as anandamide) and 2-arachidonoyl glycerol (2-AG) are hydrolysed to arachidonic acid by the enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively. Blocking these enzymes with various synthetic compounds leads to increased levels of these endocannabinoids.

View Larger
Glossary.
