(−)-Cannabidiol (CBD) is a major, non psychotropic constituent of cannabis. It has been shown to cause numerous physiological effects of therapeutic importance. We have reported that CBD derivatives in both enantiomeric series are of pharmaceutical interest. Here we describe the syntheses of the major CBD metabolites, (−)-7-hydroxy-CBD and (−)-CBD-7-oic acid and their dimethylheptyl (DMH) homologs, as well as of the corresponding compounds in the enantiomeric (+)-CBD series. The starting materials were the respective CBD enantiomers and their DMH homologs. The binding of these compounds to the CB1 and CB2 cannabinoid receptors are compared. Surprisingly, contrary to the compounds in the (−) series, which do not bind to the receptors, most of the derivatives in the (+) series bind to the CB1 receptor in the low nanomole range. Some of these compounds also bind weakly to the CB2 receptor.
Lum´ır O. Hanuˇs,*a Susanna Tchilibon,a Datta E. Ponde,a Aviva Breuer,a Ester Frideb and Raphael Mechoulam
a Department of Medicinal Chemistry and Natural Products, School of Pharmacy, Medical Faculty, The Hebrew University of Jerusalem, Ein Kerem, 91120, Jerusalem, Israel. E-mail: lumir@cc.huji.ac.il; Fax: +972-2-6757076; Tel: +972-2-6758042 b Department of Behavioural Sciences, College of Judea and Samaria, Ariel, 44837, Israel Received 8th November 2004, Accepted 25th January 2005 First published as an Advance Article on the web 16th February 2005
Introduction
Cannabidiol (CBD) (4a) is the major non psychotropic, neutral cannabinoid in most cannabis preparations, such as marijuana and hashish. It was isolated in the early 1940s,(1) but its structure and absolute configuration were fully elucidated only in the mid 1960s.(2) Several syntheses of CBD and of its (+)-enantiomer have been reported; however most of them were low yielding.(3) An improved synthesis of (−)-CBD and of its dimethylheptyl homolog (CBD-DMH)was reported by Baek et al.(4) For a review of the chemistry of CBD see reference(5). The precursor of CBD, namely cannabidiolic acid, is usually themajor cannabinoid present in the cannabis plant and CBD is actually a product formed on decarboxylation of cannabidiolic acid.(6) In cannabis preparations, such as hashish and marijuana, CBD is found in higher concentrations than in the plant, presumably due to decarboxylation during the collection and drying of the material.(7) Cannabidiolic acid was isolated and its structure was elucidated by ˇSantav´y’s group and our group.2b,(8) CBD has been shown to cause a wide range of biological effects. These observations may be of clinical importance as CBD has low toxicity and causes no psychotropic effects in either humans or animals. Thus, CBD has been found to be anti-epileptic(9) and anxiolytic(10) in man. Recently we showed that CBD and the related 7-nor-7-carboxy-CBD-DMH (18b) are potent anti-arthritic therapeutics in models of arthritis in mice.(11)a–d Other physiological effects reported are prolongation of barbiturate sleeping time, apparently caused by inhibition of barbiturate metabolism,(12) reduction of serotonin uptake,(13) prevention of vomiting and nausea,(14) extinction of memories in animal models(15) and inhibition of neurodegeneration in an animal model of Parkinson’s disease.(16) Cannabidiol is a potent anti-oxidant.(17) For a review of the biological effects of CBD see reference(18). In view of the therapeutically promising effects of CBD we assayed several derivatives both in the natural (−) series as well as in the unnatural (+) series.
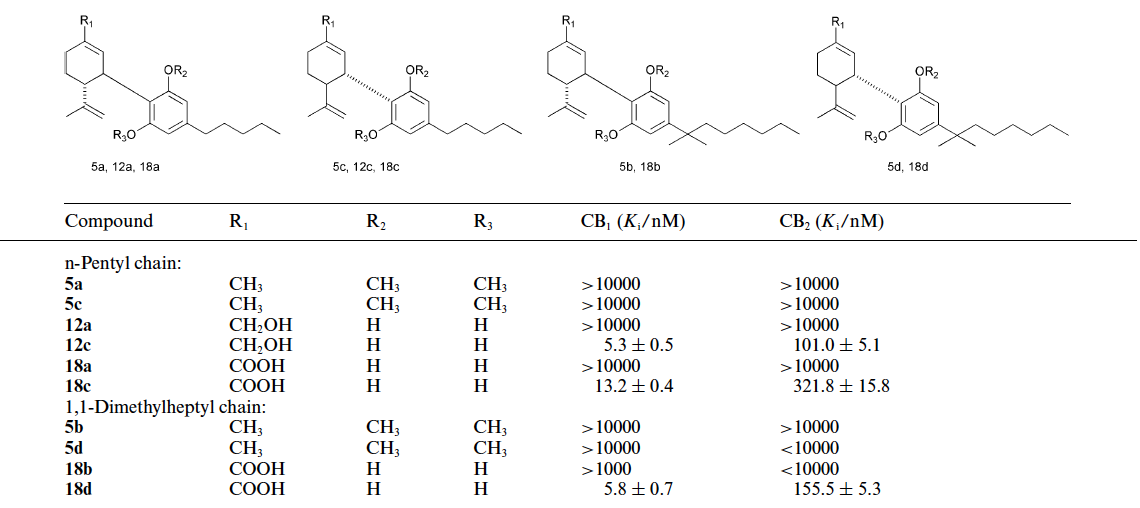
We reported that the compounds in the (−) series do not bind to the cannabinoid CB1 receptor but, surprisingly, derivatives in the (+) series bind to this receptor.(19)We also showed that some of the (+)CBDderivatives, which show significant binding to the CB1 receptor, do not exhibit any effects in the tetrad group of assays (ambulation, sedation, analgesia, temperature lowering), which are typical for cannabinoid CB1 agonists.(20) This observation indicates that these (+)-CBD derivatives apparently do not activate the CB1 receptor in the brain. Although the reason for this discrepancy is not known, such compounds may be of significant therapeutic interest as non-psychotropic cannabinoid agonists to the peripheral CB1 receptors, with possible activity in reduction of peripheral pain and inflammation. Peripherally restricted agonists to the CB1 receptor have not been described so far. In the present publication we describe the syntheses of (−)- CBD metabolites and derivatives, as well as (+)-CBD derivatives, whose binding to the CB1 receptor we have previously described,(19) and some new additional, related compounds. The binding of all new compounds are presented in Table 1, together with those of the corresponding enantiomers. The binding of all previously reported compounds and those reported here, is summarized in two Tables as Electronic Supplementary Information.† The synthetic procedures described include those of the major CBD metabolites, (−)-7-hydroxy-CBD (12a) and (−)-CBD-7-oic acid (18a) and their dimethylheptyl (DMH) homologs (12b and 18b) aswell as the corresponding compounds in the enantiomeric (+)-CBD series (12c,18c,12d and 18d). We have published a short communication on the synthesis of (−)-7-hydroxy-CBD (12a),(21) and a low yield synthesis of (+)-7-hydroxy-CBD diacetate.22 The starting materials for the syntheses of the CBD derivatives were (−)-CBD (4a) and (−)- CBD-DMH (4b) and their enantiomers (4c) and (4d).
Results and discussion
Syntheses
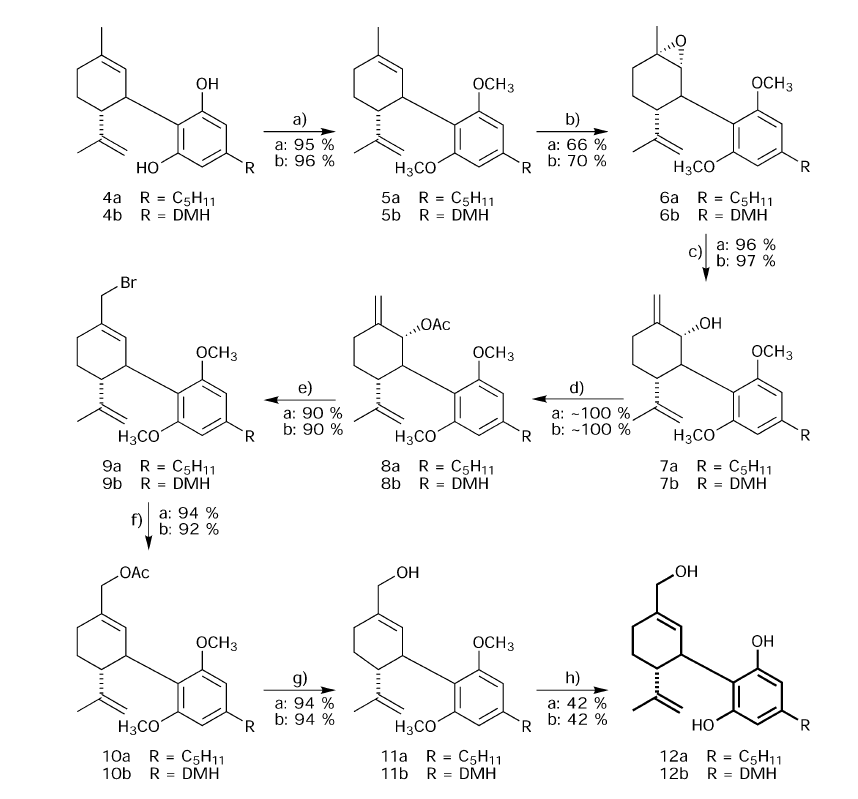
The syntheses of (+)-CBD (4c) and of (+)-CBD-DMH (4d) are depicted in Scheme 1. Our synthetic route follows that generally employed in most cannabinoid total syntheses, namely the condensation of a monoterpenoid allylic alcohol with a resorcinol derivative. Condensation of (−)-p-mentha-1,8-diene-3-ol3c with olivetol in the presence of boron trifluoride in diethyl ether led predominantly to the formation of the cyclised (+)-D9-THC (3).However when the modified procedure reported by Baek et al.4 was employed, namely condensation with boron trifluoride etherate absorbed on basic alumina, we obtained the desired 4c in 44% and 4d in 55% yields. The syntheses of 12a and 12b are depicted in Scheme 2. CBD (4a) was converted into its dimethyl ether (5a) by dimethyl sulfate–potassium carbonate in acetone, which, on reaction with one mole of meta chloroperbenzoic acid gave the epoxide (6a).
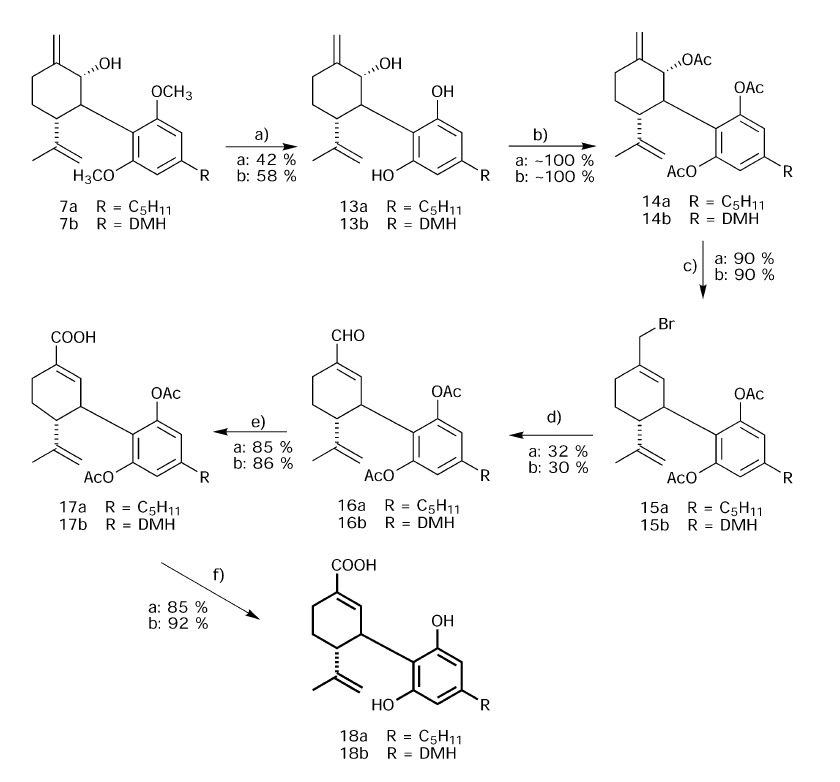
Epoxidation, being an electrophilic reaction, selectively attacked the ring double bond without a reaction on the terminal olefinic group, as the electron density on the latter is lower than in the former. The epoxide presumably is trans to the aromatic ring, on the basis of related previous NMR studies.2a Methylmagnesium-N-cyclohexylisopropylamide (prepared in situ) opened the epoxide ring, to give 7a exclusively. The use of methyl ether as a protecting group was found to be necessary. Every attempt to change it to a different group that is easier to remove, such as methoxyethoxymethyl (MEM), methoxymethyl (MOM) or silyl ethers, was unsuccessful as the epoxidation reaction did not proceed or the protecting group was removed during the reaction. Acetylation of 7a with acetic anhydride–pyridine gave 8a, which was converted by tbutyldimethylsilyl bromide (TMSBr) into the allylic bromide 9a.Reactionwith tetrabutylammonium acetate led to 10a,which gave 11a on hydrolysis. The ether blocking groups were removed by heating with methylmagnesium iodide at 200 ◦C,23 producing (−)-7-hydroxy-cannabidiol (12a), a primary CBD metabolite. The same sequence of reactions starting with (−)-CBD-DMH (4b) led to (−)-7-hydroxy-CBD-DMH (12b). The second major metabolite, CBD-7-oic acid (18a), was prepared as described in Scheme 3. Here the same reaction sequence was followed up to the allyl alcohol (7a), as in Scheme 2. Then the ether protecting groups were removed with methylmagnesium iodide at 200 ◦C, leading to the triol (13a), which was then acetylated to the triacetate (14a). On bromination, as described above, the bromide 15a was obtained. The bromide 15a was oxidized with potassium chromate in hexamethylphosphoric triamide to the aldehyde 16a, which on further oxidation with sodium chlorite led to (−)-CBD-DMH- 7-oic acid diacetate (17a). The deacetylation was carried out by sodium borohydride in ethanol to give desired acid metabolite (18a). The same pathway was followed in the synthesis of the dimethylheptyl homolog 18b.
The synthetic pathways described above represent the first preparation of the major natural and unnatural CBD metabolites, 12a and 18a, and their dimethylheptyl homologs 12b and 18b and make them available for biological evaluation. The same sequence of reactions starting from the enantiomeric (−)-p-mentha-1,8-diene-3-one led to the (+) enantiomeric alcohols 12c,18c and carboxylic acids 12d and 18d. Binding to the cannabinoid receptors CB1 and CB2 In a previous publication describing the biological properties of cannabidiol derivatives, we presented binding data for some of the compounds whose synthesis is described here, namely 4a– d,12a,b,d,18a,b.Wewere surprised to note thatwhile compounds in the natural, levorotatory, 3R,4R series did not bind, or bound very weakly, to the CB1 and the CB2 cannabinoid receptors, compounds in the dextrorotatory, 3S,4S series showed potent binding to CB1 and somewhat lower binding to CB2. Thus (−)- CBD-DMH (4b) binds to the CB1 receptor with a Ki above 10 lMand to the CB2 receptor with a Ki of 1800 nM, while the (+) enantiomer (4d) does so with a Ki of 17.4 nM and 211 nM respectively. In the 7-hydroxy series (+)-7-OH-CBD-DMH (12d) binds with a Ki of 2.5 nMto CB1 and 44.0 nMto CB2, while the numbers for the (−) enantiomer (12b)were 4400nMand 671nM respectively. When all the cannabidiol metabolites not assayed previouslywere investigatedwe observed the same phenomenon. We compared five sets of enantiomers (see Table 1). The phenolic ethers, 5a and 5c, as well as 5b and 5d, did not bind to either CB1 or CB2. In the three remaining sets in which the phenolic groups are not substituted, the (−) enantiomers (12a,18a,b) were essentially inactive on binding, while potent activity was noted with the (+) enantiomers (12c,18c,d). These stereochemical differencesmay be useful in future investigations on the structural features of the receptors which are required for binding. We would like to stress that not all cannabinoid activities are CB1/CB2-mediated. Several additional, putative receptors have been proposed but so far none of these have been cloned or well-identified.24 Indeed (−)-CBD (4a) and the acid 18b, which do not bind significantly to either receptor, are potent anti-inflammatory compounds in models of rheumatoid arthritis. The molecular mechanism of this activity is unknown. In summary, we report a synthetic pathway to the unnatural enantiomer of CBD, namely (+)-CBD (4c) and to its DMH homologue, (+)-CBD-DMH (4d), as well as the first syntheses of the major (−)-CBD metabolites (−)-7-hydroxy-CBD (12a) and (−)-CBD-7-oic acid (18a) and the corresponding compounds in the (+) series, and report their binding to the CB1 and CB2 receptors.
Experimental
General remarks
1H-NMR spectra were measured on a Varian VXR-300S spectrophotometer using CDCl3 as solvent with TMS as the internal standard. All chemical shifts are reported in ppm. Specific rotations were determined with a Perkin-Elmer 141 polarimeter. Column chromatography was performed with ICN silica gel 60 A˚ . Organic solvents were dried over anhydrous sodium sulfate.
Preparation of synaptosomal membranes and transfected cells
Synaptosomal membranes, used in this assay for CB1 receptor binding, were prepared from the brains of Sabra male rats (250– 300 g) after removal of the brain stem by centrifugation and gradient centrifugation after their homogenization.25 For CB2 receptor binding assays transfected cells were prepared. COS- 7 cells were transfected with plasmids containing CB2 receptor cDNA, and crude membranes were prepared.26
Receptor binding assays
The high affinity receptor probe,27 [3H]HU-243 (Tocris Cookson Ltd., United Kingdom), with a dissociation constant of 45 °æ 7 pM for the CB1 receptor, was incubated with synaptosomal membranes (3–4 lg) for CB1 assays and/or transfected cells for CB2 assays, for 90 min at 30 ◦C with different concentrations of the assayed CBD derivatives or with the vehicle alone (fattyacid- free bovine serum albumin at a final concentration of 0.5 mg ml−1). Bound and free radioligands were separated by . centrifugation. The data were normalized to 100% of specific binding, which was determined with 50 nM unlabeled HU-243. The results presented are the average of triplicate determination from three independent experiments. The Ki value was determined with the GraphPad Prism (Version 3.02) program which follows the Cheng–Prusoff equation. A sigmoid dose-response (variable slope) built-in equation in this Prism program was used to fit the curves.
Dimethoxy-CBD (5a)
CBD, isolated from hashish, (3 g, 9.95 mmol) was dissolved in DMF (55 ml). K2CO3 (7.35 g, 53.3 mmol) and CH3I (2.3 ml, 36.9 mmol) were added and the mixture was stirred at room temperature for 4 hours. The reaction was monitored by TLC (10% ether–petroleum ether) until the starting material had disappeared. Then water (200 ml) was added and the solution was extracted with ether. The organic phase was washed with brine until neutral, dried on MgSO4 and filtered. Removal of the solvent under reduced pressure afforded 3.2 g of the product 5a. Yield 98%; 1H-NMR: d 6.344 (2H, s, Ar), 5.220 (1H, s, olefin), 4.460–4.436 (2H, d, J = 7.2 Hz), 4.023–3.971 (1H, m, benzyl), 3.741 (6H, s, OCH3), 2.960–2.869 (1H, td, J = 11.5, 4.5 Hz, allyl), 2.717–2.569 (2H, t, J = 7.5 Hz, benzyl), 2.259–2.144 (1H, m), 2.018–1.960 (1H, m), 1.789–1.722 (1H, m), 1.678 (3H, s, allyl CH3), 1.568 (6H, br s), 1.352 (4H, m), 0.936–0.890 (3H, t, J = 6.8 Hz, terminal CH3); IR mmax/cm−1:2875, 1600, 1570, 1440, 1410, 1220, 1100, 880; [a]20D−96.8 (c12.19 mg ml−1 in CHCl3); MS m/z: 342 (M+, 14%), 274 (100),243 (27), 235 (10), 221 (40), 173 (16); HR-MS m/z calculated for C23H35O2: 342.2559, found 342.2551.
Dimethoxy-CBD-DMH (5b)
Prepared by the same procedure reported for 5a, with CBDDMHas starting material. Yield 96%; 1H-NMR: d 6.449 (2H, s, Ar), 5.238 (1H, s, olefin), 4.422–4.382 (2H, d, J = 12.0 Hz), 4.120–3.901 (1H, m, benzyl), 3.784 (6H, s, OCH3), 2.933–2.801 (1H, m, benzyl), 2.270–2.086 (1H, m, allyl), 2.048–1.924 (1H, m), 1.781–1.501 (10H, m), 1.253–1.185 (10H, m), 1.105–0.962 (2H, m), 0.849–0.8816 (3H, t, J = 6.8 Hz, terminal CH3); IR mmax/cm−1: 2900, 1600, 15780, 1440, 1400, 1100; [a]20 D−98.1 (c 2.04 mg ml−1 in CHCl3); MS m/z: 398 (M+, 19%), 331 (25), 330 (100), 301(14), 291(15), 277 (57), 245 (35); HR-MS m/z calculated for C27H42O2: 398.3185, found 398.3186.
1,6-Epoxy-2,6-dimethoxy-dihydrocannabidiol (6a)
-Chloroperbenzoic acid (70% pure 1.2 g, 4.85 mmol) was dissolved in 50 ml CH2Cl2 and the solution was cooled to 0 ◦C. A solution of 5a (1.65 g, 4.82 mmol) in 10 ml CH2Cl2 was slowly injected. The reaction mixture was stirred at 0 ◦C for 30 min and monitored by TLC (10% ether–petroleum ether). The reaction was quenched by addition of a saturated aqueous solution of NaHCO3 and the organic phase was separated by a separatory funnel, then the aqueous phase was extracted with ether. The combined organic extracts were washed with brine, dried over MgSO4 and filtered. Removal of the solvents under reduced pressure afforded a residue that was flash chromatographed (7% ether–petroleum ether) to give the epoxy-derivative 6a. Yield 65%; 1H-NMR: d 6.348–6.322 (2H, d, J = 7.7 Hz, Ar), 4.369 (1H, s, olefin), 4.159 (1H, s, olefin), 3.803 (3H, s, OCH3), 3.714 (3H, s, OCH3), 3.612–3.571 (1H, d, J = 12.2, Hz, H on epoxide ring), 2.574–2.522 (2H, t, J = 7.9 Hz, benzyl), 2.293–2.201 (1H, m), 2.081–1.995 (1H, m), 1.882–1.757 (1H, m), 1.628–1.585 (6H, m), 1.364–1.313 (9H, m), 0.936–0.890 (3H, t, J = 6.5 Hz, terminal CH3); IR mmax/cm−1: 2900, 1610, 1580, 1460, 1420, 1120, 760; MS m/z: 358 (M+, 26%), 341 (5), 287 (7), 274 (16), 250 (29), 221 (100); HR-MS m/z calculated for C23H34O3: 358.2508, found 358.2531. 1,6-Epoxy-2,6-dimethoxy-dihydrocannabidiol-DMH (6b) Prepared by the same procedure as reported above for 6a. Yield 70%; 1H-NMR: d 6.466–6.442 (2H, d, J = 7.2 Hz, Ar), 4.358 (1H, s, olefin), 4.121 (1H, s, olefin), 3.805 (3H, s, OCH3), 3.719 (3H, s, OCH3), 3.591–3.555 (1H, d, J = 10.8, Hz, H on epoxidering), 2.235–2.193 (1H, m, benzyl), 2.105–1.995 (1H, m, allyl), 1.907–1.761 (1H, m), 1.745–1.514 (10H, m), 1.369 (3H, s, allyl CH3), 1.268–1.180 (10H, m), 1.081–0.942 (2H, m.), 0.856–0.812 (3H, t, J = 6.5 Hz, terminal CH3); IR mmax/cm−1: 2900, 1600, 1580, 1460, 1450, 1210, 1110, 750; MS m/:414 (M+, 13%), 346 (26), 331 (13), 290 (80), 277 (100), 261 (20), 221 (21); HR-MS m/z calculated for C27H42O3: 414.3134, found 414.3098.
(3R,4R)-3-(4-Pentyl-2,6-dimethoxyphenyl)-2-hydroxy-4- isopropenyl-1-methylenecyclohexane (7a)
Butyllithium in hexane (5.6ml, 14 mmol)was added to a solution of N-cyclohexylisopropylamine (1.85 ml, 11.3 mmol) at 0 ◦C in anhydrous toluene (10 ml, distilled over sodium) under an N2 atmosphere. After 15 min, methylmagnesium bromide in ether (3.8 ml, 11.4 mmol) was injected, and the reaction mixture was stirred for 45 min at room temperature. A solution of 6a (1 g, 2.79 mmol) in dry toluene (3 ml) was added, and the mixture was heated to 40 ◦C and stirred for two hours. Then the reaction was cooled to 0 ◦C and quenched by the slow addition of 5 M HCl. The organic phase was separated by a separatory funnel, and the aqueous phase was extracted with ether. The combined organic extracts were washed with brine, dried over MgSO4 and filtered. Removal of the solvents under reduced pressure afforded a residue that on TLC (20% ether–petroleum ether) showed only one spot, and by 1H-NMR was identified as 7a. Yield 97%; 1H-NMR: d 6.332 (2H, s, Ar), 5.083 (1H, s, olefin), 4.821 (1H, s, olefin), 4.662–4.622 (1H, d, J = 11.8 Hz, CHOH), 4.387 (1H, s, olefin), 4.379 (1H, s, olefin), 3.798 (3H, s, OCH3), 3.745 (3H, s, OCH3), 3.200–3.154 (1H, td, J = 11.2, 3.0 Hz, benzyl), 2.564–2.452 (3H, m), 2.255–1.625 (1H, m), 1.754–1.707 (1H, m), 1.609–1.350 (4H, m), 1.432 (3H, s, allyl CH3), 1.350– 1.313 (4H, m), 0.924–0.878 (3H, t, J =6.5Hz, terminalCH3); IR mmax/cm−1: 3400, 2920, 1590, 1450, 1120, 900, 730; [a]20D+62.3 (c15.36 mg ml−1 in CHCl3); HR-MS m/z calculated for C23H34O3:358.2508, found 358.2508.
(3R,4R)-3-[4-(1,1-Dimethylheptyl)-2,6-dimethoxyphenyl]-2-
hydroxy-4-isopropenyl-1-methylenecyclohexane (7b) Prepared by the same procedure as reported above for 7a. Yield 97%; 1H-NMR: d 6.440 (2H, s, Ar), 5.080 (1H, s, olefin), 4.821 (1H, s, olefin), 4.655–4.621 (1H, d, J = 9.0 Hz, CHOH), 4.448 (1H, s, olefin), 4.338 (1H, s, olefin), 3.802 (3H, s, OCH3), 3.744 (3H, s, OCH3), 3.215–3.127 (1H, td, J = 11.7, 3.0 Hz, benzyl), 2.505–2.444 (1H, dt, J = 12.6, 3.0 Hz allyl), 2.255–2.182 (1H, td, J = 9.0, 3.0 Hz), 1.740–1.688 (2H, m), 1.555–1.423 (8H, m), 1.301–1.177 (10H, m), 1.025–0.955 (2H, m), 0.859–0.814 (3H, t, J =6.5Hz, terminalCH3); IR mmax/cm−1: 3400, 2900, 1600, 1560, 1450, 1400, 1110, 750; [a]20D +47.6 (c 1.05 mg ml−1 in CHCl3); MS m/z: 414 (M+, 10%), 370 (45), 290 (22), 278 (20), 277 (100); HR-MSm/z calculated forC27H42O3: 414.3134, found 414.3134.
(3R,4R)-3-[2,6-Dimethoxy-4-pentylphenyl]-2-acetoxy-
4-isopropenyl-1-methylenecyclohexane (8a) 7a (0.9 g, 2.5 mmol) was dissolved in pyridine (2 ml) and acetic anhydride (2 ml) and the reaction was stirred for 18 hours at room temperature. Then the solution was poured onto iced water (20 ml) and extracted with ether. The combined organic extracts were washed successively with 1 M HCl, aqueous sodium bicarbonate and brine, dried on MgSO4 and filtered. Removal of the solvents under reduced pressure afforded an oily residue that on TLC (20% ether–petroleum ether) showed only one spot, that by 1H-NMR was proved to be 8a. Yield ∼100%; 1H-NMR: d 6.281–6.267 (2H, d, J = 4.2 Hz, Ar), 5.967–5.931 (1H, d, J = 10.8 Hz, olefin), 4.767–4.721 (2H, d, J = 13.7 Hz,olefin), 4.535 (1H, s, olefin), 4.419 (1H, s, olefin), 3.793 (3H, s, OCH3), 3.745 (3H, s, OCH3), 3.491–3.416 (1H, t, J = 11.4 Hz), .286–3.197 (1H, td, J = 11.4, 2.7, Hz, benzyl), 2.533–2.469 (2H, t, J = 7.2 Hz), 2.325–2.249 (1H, m), 1.717 (3H, s, OAc), 1.625–1.447 (6H, m), 1.404–1.250 (6H, m), 0.924–0.878 (3H, t, J = 6.5 Hz, terminal CH3); IR mmax/cm−1: 2910, 1750, 1450, 1360, 1240, 1120, 890; MS m/z: 400 (M +, 16%), 340 (14), 314 (73), 234 (22), 221 (100); HR-MS m/z calculated for C25H36O4: 400.2614, found 400.2603.
(3R,4R)-3-[4-(1,1-Dimethylheptyl)-2,6-dimethoxyphenyl]-2-
acetoxy-1-methylenecyclohexane (8b) Prepared by the same procedure as reported above for 8a. Yield ∼ 100%; 1H-NMR: d 6.409–6.377 (2H, d, J = 8.1 Hz, Ar), 5.980–5.931 (1H, d, J = 14.5 Hz, CHOAc), 4.768–4.717 (2H, d, J = 15.2 Hz, olefin), 4.521 (1H, s, olefin), 4.405 (1H, s, olefin), 3.802 (3H, s, OCH3), 3.754 (3H, s, OCH3), 3.268–3.181 (1H, m, benzyl), 2.522–2.459 (1H, m, allyl), 1.781–1.717 (1H, m), 1.695 (3H, s,OAc), 1.540–1.484 (6H, m), 1.239–1.171 (14H,m), 0.980– 0.923 (2H, m), 0.854–0.809 (3H, t, J = 6.7 Hz, terminal CH3); IR mmax/cm−1: 290, 1750, 1450, 1360, 1240, 1120, 880; MS m/z: 456 (M+, 40%), 396 (11), 370 (100), 290 (28), 277 (41); HR-MS m/z calculated for C29H44O4: 456.3239, found 456.3222.
7-Bromo-dimethoxy-CBD (9a)
8a (1 g, 2.5 mmol) was dissolved in dry CH2Cl2 (50 ml, distilled over CaH2) under nitrogen atmosphere and TMSBr (1.6 ml, 12.1 mmol) was added. The reaction was stirred at rt for 4 hours, then it was shaken with a saturated aqueous solution of NaHCO3 and the organic phase was separated by a separatory funnel, and the aqueous phase was extracted with ether. The combined organic extracts were washed with brine, dried over MgSO4 and filtered. Removal of the solvents afforded a residue that 1H-NMR and TLC (20% ether–petroleum ether) showed predominantly a single component, that was used with no purification. Yield 90%; 1H-NMR: d 6.322 (2H, s, Ar), 5.736 (1H, s, olefin), 4.767 (1H, s, olefin), 4.454), 4.535 (1H, s, olefin), 4.006 (2H, s, CH2Br), 3.736 (6H, s, OCH3), 2.853–2.767 (1H, td, J = 11.9, 3.2 Hz, benzyl), 2.565–2.512 (1H, t, J = 7.9, Hz, benzyl), 2.397–2.359 (1H, m), 2.277–2.183 (1H, m), 1.870–1.662 (2H, m), 1.619 (3H, s, allyl CH3), 1.439–1.237 (7H, m), 0.928– 0.882 (3H, t, J = 6.6 Hz, terminal CH3); IR mmax/cm−1: 2900, 1580, 1460, 1230, 1120; [a]20D −20.5 (c 1.7 mg ml−1 in ethanol); MS m/z): 423 (M+, 0.6%), 342 (27), 340 (28), 287 (42), 274 (61), 221 (100); HR-MS m/z calculated for C23H34O2 Br: 423.1722, found 423.1708.
7-Bromo-dimethoxy-CBD-DMH (9b)
Prepared by the same procedure as reported above for 9a. Yield 90%; 1H-NMR: d 6.431 (2H, s, Ar), 5.602 (1H, s, olefin), 4.821–4.337 (4H, m, CH2Br + olefin), 4.042–3.961 (1H, m, olefin), 3.720 (6H, s, OCH3), 3.116–3.010 (1H, m, benzyl), 2.842–2.762 (1H, allyl), 1.782–1.517 (9H, m), 1.247–1.178 (10H, m), 1.010 (2H, br s), 0.831 (3H, br s, terminal CH3); IR mmax/cm−1: 2910, 1580, 1460, 1230, 1120; [a]20D−17.5 (c 6.1 mg ml−1 in ethanol); HR-MS m/z calculated for C27H42O2Br: 477.2368, found 477.2378.
7-Acetoxy-dimethoxy-CBD (19a)
9a (570 mg, 1.35 mmol) was dissolved in acetone (15 ml, stored on 4 A˚ molecular sieves) and tetrabutylammonium acetate (450 mg, 1.49 mmol) was added. The mixture was stirred, refluxed and monitored by TLC (20% ether–petroleum ether). After 2 hours there was no more starting material. The acetone was removed under reduced pressure, and the residuewas diluted with water (20 ml) and extracted with ether. The combined organic extracts were washed with aqueous sodium bicarbonate and brine, dried on MgSO4 and filtered. Removal of the solvents under reduced pressure afforded 520 mg of an oily residue. Yield 96%; 1H-NMR: d 6.320 (2H, s, Ar), 5.581 (1H, s, olefin), 4.492–4.386 (4H, m, CH2OAc + olefin), 4.040–3.986 (1H, m, benzyl), 3.715 (6H, s, OCH3), 2.853–2.801 (1H, m), 2.195–2.071 (2H, m), 2.060 (3H, s, OAc), 1.823–1.695 (2H, m), 1.605 (5H, br s), 1.323 (4H, br s), 0.921–0.875 (3H, t, J = 6.7 Hz, terminal CH3); IR mmax/cm−1: 2900, 1720, 1580, 1440, 1110; [a]20D−135.2 (c 15.95 mg ml−1, CHCl3); MS m/z: 400 (M+, 3%), 332 (26), 331 (100), 241 (41), 221 (55), 208 (11); HR-MS m/z calculated for C25H36O4: 400.2614, found 400.2609.
7-Acetoxy-dimethoxy-CBD-DMH (10b)
Prepared by the same procedure as reported above for 10a, but the yield was slightly lower. Yield 90%; 1H-NMR: d 6.440 (2H, s, Ar), 5.609 (1H, s, olefin), 4.498–4.343 (4H, m,CH2OAc+olefin), 4.041–3.965 (1H, m, benzyl), 3.719 (6H, s, OCH3), 2.845–2.763 (1H, m, allyl), 2.193–2.099 (2H, m), 2.061 (3H, s, OAc), 1.796– 1.776 (2H, m), 1.594–1.518 (7H, m), 1.254–1.179 (10H,m), 1.015 (2H, br s), 0.856–0.861 (3H, t, J = 6.4 Hz, terminal CH3); IR mmax/cm−1: 2900, 1720, 1600, 1580, 1450, 1410, 1220; [a]20D−90.5 (c 2.53 mg ml−1, CHCl3); MS m/z: 456 (M+, 7%), 396 (8), 388 (71), 383 (25), 303 (13), 277 (68); HR-MS m/z calculated for C29H44O4: 456.3239, found 456.3239.
7-Hydroxy-dimethoxy-CBD (11a)
10a (500 mg, 1.25 mmol) was dissolved in ethanol (20 ml), 1 M NaOH (2 ml) was added and the reaction was refluxed for 1 hour. The ethanol was removed under reduced pressure, and the residue was diluted with water (20 ml) and 2 M HCl was added till acidic. The solution was extracted with ether. The combined organic extracts were washed with brine, dried on MgSO4 and filtered. Removal of the solvents under reduced pressure afforded 430 mg of an oily residue. Yield 96% yield; 1H-NMR: d 6.328 (2H, s, Ar), 5.510 (1H, s, olefin), 4.458–4.414 (2H, d, J = 13.2 Hz, olefin), 4.010 (2H, br s, CH2OH), 3.728 (6H, s, OCH3), 2.858–2.806 (1H, m, benzyl), 2.566–2.508 (2H, t, J = 7.5 Hz, benzyl), 2.213 (2H, m), 1.817–1.582 (7H, m), 1.451– 1.259 (5H, m), 0.924–0.878 (3H, t, J = 6.5 Hz, terminal CH3); IR mmax/cm−1: 3300, 2900, 1580, 1440, 1220, 1110; [a]20D −80.7 (19 mg/10 ml ethanol); MS m/z: 358 (M+, 7%), 327 (52), 290 (80), 221 (100), 152 (33); HR-MS m/z calculated for C25H38O3: 358.25080, found 358.2511.
7-Hydroxy-dimethoxy-CBD-DMH (11b)
Prepared by the same procedure as reported above for 11a.Yield 94%; 1H-NMR: d 6.446 (2H, s, Ar), 5.528 (1H, s, olefin), 4.434– 4.367 (2H, d, J = 20.1 Hz, olefin), 4.010 (3H, br s, CH2OH + OH), 3.729 (6H, s, OCH3), 2.905–2.785 (1H, m, benzyl), 2.248– 2.105 (2H, m), 1.759–1.704 (2H, m), 1.535 (3H, s, allyl CH3), 1.495–1.460 (4H, m), 1.360–1.120 (10H, m), 0.980–0.9875 (2H, m), 0.797–0.752 (3H, t, J =6.5Hz, terminalCH3); IR mmax/cm−1: 3300, 2900, 1600, 1570, 1420, 1400, 1230, 1110, 750; [a]D −135.2 (c 15.95 mg ml−1, CHCl3); MS m/z: 414 (M+, 14%), 396 (8), 383 (100), 346 (43), 277 (50), 119 (7); HR-MS m/z calculated for C27H42O3: 414.3134.
7-Hydroxy-CBD (12a)
A Grignard reagent was prepared with magnesium (100 mg, 4.17 mmol) and CH3I (0.26 ml, 4.17 mmol) in dry ether (3 ml, distilled over sodium) under N2 atmosphere. 11a (420 mg, 1.17 mmol) in ether (1 ml) was slowly added to the stirred solution and the ether was distilled off. The residue was heated under N2 atmosphere to 210 ◦C for 45 min. The flask was cooled to room temperature and the reaction was quenched with ice water. The aqueous solution was extracted with ether several times. The combined organic extracts were dried on MgSO4 and filtered. Removal of the solvents under reduced pressure afforded a residue that was chromatographed on silica gel (25% ether–petroleum ether) to give 150 mg of pure 12a. Yield 40%); 1H-NMR: d 6.200 (2H, s, Ar), 5.822 (1H, s, olefin), 4.629 (1H, s, olefin), 4.518 (1H, s, olefin), 4.075 (2H, s, CH2OH), 3.962–3.923 (1H, m, benzyl), 2.567–2.484 (1H, td, J = 13.3, 2.7 Hz, allyl), 2.435–2.384 (2H, t, J = 7.5 Hz, benzyl), 1.882– 1.734 (2H, m), 1.660 (6H, s. allyl CH3), 1.584–1.487 (2H, m), 1.285–1.248 (6H, m), 0.886–0.843 (3H, t, J = 6.3 Hz, terminal CH3); IR mmax/cm−1: 3300, 2900, 1620, 1580, 1440, 1240, 1020, 730; [a]D −67.3 (c 19.51 mg ml−1, CHCl3 ); MS m/z: 330 (M+, 10%), 312 (44), 299 (53), 284 (44), 244 (100), 231(56), 187 (29), 147 (13);HR-MSm/z calculated forC21H30O3: 330.21949, found 330.2231.
7-Hydroxy-CBD-DMH (12b)
Prepared by the same procedure as reported above for 12a.Yield 42%; 1H-NMR: d 6.335 (2H, s, Ar), 5.863 (1H, s, olefin), 4.652 (1H, s, olefin), 4.538 (1H, s, olefin), 4.108 (2H, s,CH2OH), 3.920– 3.889 (1H, d, J = 9.3 Hz, benzyl), 2.498–2.433 (1H, m, allyl), 2.228 (2H, br s), 2.064–1.715 (2H, m), 1.648–1.428 (7H, m), 1.312–1.168 (12H, m), 0.853–0.808 (3H, t, J = 6.5 Hz, terminal CH3); IR mmax/cm−1: 3300, 2900, 1620, 1580, 1420, 1210, 1020, 750; [a]D −61.1 (c 1.8mgml−1, CHCl3);MSm/z: 386 (M+, 24%), 369 (30), 368 (30), 355 (100), 300 (43), 287 (510), 283 (34), 249 (38), 233 (22), 187 (10); HR-MS m/z calculated for C25H38O3: 386.28210, found 386.2825.
(3R,4R)-3-[2,6-Dihydroxy-4-pentylphenyl]-2-hydroxy-4-
isopropenyl-1-methylenecyclohexane (13a) A Grignard reagent was prepared with magnesium (84 mg, 3.5 mmol) and CH3I (0.2 ml, 3.5 mmol) in dry ether (1 ml, distilled over sodium) underN2 atmosphere. 7a (360 mg, 1 mmol) in ether (0.5 ml) was added to the stirred solution and the ether was distilled. The residue was heated under N2 atmosphere to 210 ◦C for 45 min. The flask was cooled to the room temperature and the reaction was quenched with ice water. The aqueous solution was extracted several times with ether. The combined organic extracts were dried on MgSO4 and filtered. Removal of the solvents under reduced pressure afforded a residue that was chromatographed on silica gel (25% ether–petroleum ether) to give 132 mg of the pure 13a.Yield 45%; 1H-NMR: d 6.156–6.097 (2H, d, J = 17.7 Hz, Ar), 5.612 (1H, s, OH), 5.370 (1H, s, OH), 5.092 (1H, s, olefin), 4.847 (1H, s, olefin), 4.684–4.625 (2H, m, CHOH + olefin), 4.462 (1H, s, olefin), 3.300–3.205 (1H, td, J = 12.7, 2.7 Hz, benzyl), 3.128–3.058 (1H, t, J = 10.5, Hz, allyl), 2.270–2.141 (1H, m), 2.122–2.049 (1H, br s, OH), 1.767–1.712 (1H, m), 1.534–1.48 (5H, m), 1.290–1.183 (4H, m), 0.895–0.881 (3H, t, J = 6.6 Hz, terminal CH3); IR mmax/cm−1: 3350, 2900, 1620, 1580, 1420, 1160, 1000, 750; MS m/z: 330 (M+, 18%), 312 (23), 286 (14), 244 (16), 207 (27), 193 (100); HR-MS m/z calculated for C21H30O3: 330.2195, found 330.2206.
(3R,4R)-3-[4-(1,1-Dimethylheptyl)-2,6-dihydroxyphenyl]-2-
hydroxy-4-isopropenyl-1-methylenecyclohexane (13b) Prepared by the same procedure as reported above for 13a. Yield 58%; 1H-NMR: d 6.295 (1H, s. Ar), 6.229 (1H, s, Ar), 5.786 (1H, s, OH), 5.546 (1H, s, OH), 5.127 (1H, s, olefin), 4.861 (1H, s, olefin), 4.751–4.716 (1H, d, J = 3.3 Hz, CHOH), 5.127 (1H, s, olefin), 4.444 (1H, s, olefin), 3.421–3.276 (1H, m, benzyl), 3.132–3.062 (1H, t, J = 10.5, Hz, allyl), 2.502–2.459 (1H, d, J = 12.9 Hz), 2.251–2.175 (2H, m), 1.780–1.739 (1H, m), 1.528 (3H, s, allyl CH3) 1.460–1.433 (4H, m), 1.251–1.170 (10H, m), 0.954 (2H, br s), 0.845 (3H, br s, terminal CH3); IR mmax/cm−1: 3300, 2900, 1620, 1580, 1410, 1210, 750; [a]20 D +47.3 (c 1.48 mg ml−1 in CHCl3); MS m/z: 386 (M+, 60%), 368 (58), 302 (47), 283 (72), 263 (37), 262 (70), 249 (100); HR-MS m/z calculated for C25H38O3: 386.2821, found 386.278.
(3R,4R)-3-[2,6-Diacetoxy-4-pentylphenyl]-2-acetoxy-4-
isopropenyl-1-methylenecyclohexane (14a) 13a (100 mg, 0.3 mmol) was dissolved in pyridine (0.5 ml) and acetic anhydride (0.5 ml) and the reaction was stirred for 18 hours at room temperature. Then the solution was poured onto iced water (10 ml) and extracted with ether. The combined organic extracts were washed successively with 1 M HCl, aqueous sodium bicarbonate and brine, dried on MgSO4 and filtered. Removal of the solvents under reduced pressure afforded 136 mg of an oily residue that was proved to be 14a by NMR. Yield ∼100%; 1H-NMR: d 6.861 (1H, s, Ar), 6.696 (1H, s, Ar), 5.725–5.688 (1H, d, J = 11.1 Hz, CHOAc), 4.083 (1H, s, olefin), 4.689 (1H, s, olefin), 4.540–4.515 (2H, d, J = 7.5 Hz, olefin), 3.180–3.105 (1H, t, J = 11.3 Hz, benzyl), 2.893– 2.802 (1H, td, J = 11.3, 3.2 Hz, allyl), 2.563–2.513 (2H, t, J = 7.5, Hz, benzyl), 2.374 (3H, s, OAc), 2.280 (3H, s, OAc), 1.798 (3H, s, OAc), 1.614–1.470 (5H, m), 1.286–1.246 (8H, m), 0.886– 0.844 (3H, t, J = 6.3 Hz, terminal CH3); IR mmax/cm−1: 2910, 1750, 1410, 1350, 1180, 1130, 890; HR-MS m/z calculated for C27H36O6: 456.2512, found 456.2502.
(3R,4R)-3-[2,6-Diacetoxy-4-(1,1dimethylheptyl)phenyl]-2-acetoxy-4-isopropenyl-1-methylenecyclohexane (14b)
Prepared by the same procedure as reported above for 14a.Yield ∼100%; 1H-NMR: d 6.947 (1H, s, Ar), 6.795 (1H, s, Ar), 5.732– 5.695 (1H, d, J = 11.0 Hz, CHOAc), 4.798 (1H, s, olefin), 4.691 (1H, s, olefin), 4.540–4.515 (2H, d, J = 7.5 Hz, olefin), 3.167– 3.095 (1H, t, J = 11.3 Hz, benzyl), 2.854–2.816 (1H, m, allyl), 2.561–2.515 (1H, d, J = 13.8, Hz, benzyl), 2.372 (3H, s, OAc), 2.287 (3H, s, OAc), 2.230–2.195 (1H, m), 1.825–1.770 (4H, m), 1.538–1.424 (6H, m), 1.224–1.151 (12H, m), 0.955–0.945 (2H, m), 0.840–0.799 (3H, t, J =6.1Hz, terminalCH3); IR mmax/cm−1: 2900, 1750, 1410, 1360, 1180, 1130, 890; MS m/z: 512 (M+, 26%), 452 (22), 424 (24), 410 (100), 368 (16), 342 (12), 325 (32), 249 (30); HR-MS m/z calculated for C31H44O6: 512.3128, found 512.3188.
7-Bromo-diacetate-CBD (15a)
14a (100 mg, 0.2 mmol) was dissolved in dry CH2Cl2 (10 ml, distilled over CaH2) under nitrogen atmosphere. TMSBr (0.13 ml, 1 mmol) and ZnI2 (3.4 mg, 0.01 mmol) were added. The reaction was stirred at rt for 4 hours, then it was shaken with a saturated aqueous solution of NaHCO3 and the organic phase was separated by a separatory funnel. Then the aqueous phase was extracted with ether. The combined organic extracts were washed with brine, dried over MgSO4 and filtered. Removal of the solvents afforded a residue that showed only one spot onTLC (5% ether–petroleum ether) and it was used immediately with no purification. Yield 90%; 1H-NMR: d 6.764 (2H, s, Ar), 5.456 (1H, s, olefin), 4.901 (1H, s, olefin), 4.752 (1H, s, olefin), 3.930– 3.903 (2H, m, CH2Br), 3.784–3.756 (1H, d, J = 8.2 Hz, benzyl), 2.592–2.643 (2H, m,), 2.306 (6H, s, OAc), 2.198–2.131 (2H, t, J = 10.2 Hz), 1.708 (3H, s, allyl CH3), 1.698–1.472 (4H, m), 1.439–1.194 (5H, m), 0.090–0.865 (3H, t, J = 5.3 Hz, terminal CH3); IR mmax/cm−1: 2900, 1750, 1360, 1200, 1020, 900, 720; MS m/z: 478 (M+, 3%), 397 (57) 355 (96), 354 (61), 313 (100), 245 (77); HR-MS m/z calculated for C25H33O4 81Br: 478.1542, found 478.1560.
7-Bromo-diacetate-CBD-DMH (15b)
Prepared by the same procedure as reported above for 15a.Yield 90%; 1H-NMR: d 6.816 (2H, s, Ar), 5.645 (1H, s, olefin), 4.557 (1H, s, olefin), 4.448 (1H, s, olefin), 4.016–3.966 (2H, m,CH2Br), 3.483–3.405 (1H, m, benzyl), 2.655–2.459 (1H, m, allyl), 2.220 (6H, s, OAc), 1.883–1.637 (4H, m), 1.510 (3H, s, allyl CH3), 1.485–1.426 (4H, m), 1.410–1.176 (10H, m), 1.010–0.995 (2H, m) 0.853–0.807 (3H, t, J = 6.5Hz, terminal CH3); IR mmax/cm−1: 2900, 1750, 1370, 1220, 1020, 900, 750; MS m/z: 534 (M+, 9%), 489 (14), 411 (100), 393 (25), 370 (15), 351 (30), 343 (32), 301 (47), 285 (25), 283 (43), 243(24); HR-MS m/z calculated for C29H41O4Br: 532.2188, found 532.2201.
7-Nor-7-formyl-diacetate-CBD (16a)
15a (100 mg, 0.21 mmol), 18-Crown-16 (55.4 mg, 0.21 mmol) and K2CrO4 (50.9 mg, 0.26 mmol) were dissolved in anhydrous HMPA (2 ml, distilled under vacuum and stored over 4 A˚ molecular sieves). The mixture was stirred and heated at 110 ◦C for 2 hours. The reaction was cooled and quenched by addition of 1 M HCl and the aqueous phase was extracted with ether. The organic phasewaswashedwith brine, dried overMgSO4 and filtered. Removal of the solvent under reduced pressure afforded a residue that was chromatographed on silica gel (20% ether– petroleum ether) to give 30mg of pure 16a.Yield 35%; 1H-NMR: d 9.434 (1H, s, CHO), 6.778 (2H, s, Ar), 6.638 (1H, s, olefin), 4.633 (1H, s, olefin), 4.489 (1H, s, olefin), 3.746–3.718 (1H, d, J =8.4Hz, benzyl), 2.686–2.552 (4H, m), 2.304–2.075 (6H, br s), 1.965–1.921 (1H, m), 1.754–1.590 (6H, m), 1.318–1.305 (5H, m), 0.909–0.865 (3H, t, J = 6.2 Hz, terminal CH3); IR mmax/cm−1: 2900, 1750, 1670, 1160, 1020; [a]20 D −111.5 (c 3.5 mg ml−1 in CHCl3);MSm/z: 412(M+, 11%), 383 (15), 341 (30), 328 (20), 302 (37), 284 (11), 260 (100); HR-MS m/z calculated for C25H32O5: 412.2250, found 412.2263.
7-Nor-7-formyl-diacetate-CBD-DMH (16b)
Prepared by the same procedure reported for 16a. Yield 40%; 1H-NMR: d 9.420 (1H, s CHO), 6.861 (2H, s, Ar), 6.501 (1H, s, olefin), 4.611 (1H, s, olefin), 4.455 (1H, s, olefin), 3.705–3.671 (1H, m, benzyl), 2.667–2.552 (3H, m), 2.292–2.071 (6H, br s, OAc), 1.960–1.890 (2H, m), 1.601 (3H, s, allylCH3), 1.590–1.485 (4H, m), 1.241–1.711 (8H, m) 1.100–0.931 (2H, m) 0.854–0.865 (3H, t, J = 5.7 Hz, terminal CH3); IR mmax/cm−1: 2900, 1750, 1660, 1160, 1020; [a]20 D −85.7 (c 1.4mgml−1 in CHCl3); MS m/z: 468 (M+, 72%), 382 (35), 358 (40), 316 (94), 302 (11), 249 (30); HR-MSm/z calculated forC29H41O5: 468.2876, found 468.2878.
7-Nor-7-carboxy-diacetate-CBD (17a)
NaClO2 (80% pure 82.6 mg, 0.73 mmol) was added in small quantities to a stirred mixture of 16a (70 mg, 0.17 mmol), 2- methyl-2-butene (0.45 ml, 4.25 mmol) and a saturated aqueous solution of KH2PO4 (0.2 ml) in t-butanol (4 ml). The reaction was stirred at room temperature for 5 hours, and monitored by TLC (50% ether–petroleum ether). Water was added (20 ml) and the mixture was extracted several times with ethyl acetate. The organic phase was washed with brine, dried over MgSO4 and filtered. Removal of the solvent under reduced pressure afforded a residue that was chromatographed on silica gel (30% ether–petroleum ether) to give 61.8 mg of the 17a. Yield 85%; 1H-NMR: d 6.939 (1H, s, olefin), 6.770 (2H, s, Ar), 4.611 (1H, s, olefin), 4.462 (1H, s, olefin), 3.618–3.718 (1H, m, benzyl), 2.589– 2.538 (3H, m, allyl + benzyl), 2.212 (6H, s, OAc), 1.961–1.862 (1H, m), 1.858–1.641 (1H, m), 1.592 (5H, br s), 1.321–1.255 (7H, m), 0.903–0.858 (3H, t, J =6.8Hz, terminalCH3); IR mmax/cm−1: 3300, 2900, 1750, 1270, 1020; MS m/z: 428 (M+, 3%), 410 (8), 368 (43), 326 (24), 298 (14), 276 (100), 258 (24); [a]20 D −112.2 (c 3.7 mg ml−1, CHCl3); HR-MS m/z calculated for C25H32O6: 428.2199, found 428.2198.
7-Nor-7-carboxy-diacetate-CBD-DMH (17b)
Prepared by the same procedure reported for 17a. Yield 86%; 1H-NMR: d 6.946 (1H, s, olefin), 6.854 (2H, s, Ar), 4.592 (1H, s, olefin), 4.436 (1H, s, olefin), 3.635–3.590 (1H, m, benzyl), 2.605– 2.455 (1H, m, allyl), 2.208 (6H, s, OAc), 1.950–1.803 (2H, m), 1.795–1.610 (2H, m), 1.574 (3H, s, allyl CH3), 1.529–1.475 (4H, m), 1.267–1.174 (10H, m), 1.022 (2H, br s), 0.845–0.805 (3H, t, J = 6.6 Hz, terminal CH3); IR mmax/cm−1: 3300, 2900, 1750, 1270, 1020; MS m/z: 484 (M+, 9%), 466 (21), 442 (20), 424 (90), 382 (28), 374 (41), 328 (31), 314 (28), 291 (27), 247 (44); [a]20 D −122.7 (c 2.77 mg ml−1, CHCl3 ); HR-MS m/z calculated for C29H40O6: 484.2815, found 484.2792.
7-Nor-7-carboxy-CBD (18a)
17a (50mg, 0.12 mmol) was dissolved in ethanol (10ml),NaBH4 (6 mg, 0.16 mmol) was added and the reaction was refluxed for 1 hour. The ethanol was removed under reduced pressure, the residue was diluted with water (20 ml) and the solution was extracted with ether. The combined organic extracts were washed brine, dried on MgSO4 and filtered. Removal of the solvents under reduced pressure afforded a residue that was chromatographed on silica gel (30% ether–petroleum ether) to give 38.2 mg of the 18a. Yield 95%; 1H-NMR: d 7.085 (1H, s, olefin), 6.173 (2H, s, Ar), 4.604–4.566 (2H, d, J = 11.4 Hz, olefin), 4.115–4.033 (1H, m, benzyl), 2.799–2.688 (1H, m, allyl), 2.623–2.541 (1H, m), 2.444–2.391 (2H, t, J = 7.5 Hz), 1.950– 1.869 (1H, m), 1.803–1.669 (5H, m), 1.623–1.453 (4H, m), 1.309– 1.178 (5H, m), 0.902–0.857 (3H, t, J = 6.5 Hz, terminal CH3); IR mmax/cm−1: 3350, 2950, 1700, 1440, 1400, 1160, 920, 740; [a]20 D −112.3 (c 1.87 mg ml−1 in MeOH); MS m/z: 344 (M+, 11%), 299 (15), 276 (100), 220 (24), 207 (11); HR-MS m/z calculated for C21H28O4: 344.1987, found 344.1948.
7-Nor-7-carboxy-CBD-DMH (18b)
Prepared by the same procedure reported for 18a.Yield 92%; 1HNMR: d 7.121 (1H, s, olefin), 6.291 (2H, s, Ar), 4.619–4.555 (2H, d, J = 19.1 Hz, olefin), 4.036–4.033 (1H, d, J = 8.9 Hz, benzyl), 2.718–2.567 (2H, m), 2.378–2.274 (1H, m), 1.948–1.904 (1H, m), 1.828–1.765 (1H, m), 1.648 (3H, s, allyl CH3) 1.622–1.430 (4H, m), 1.236–1.189 (8H, m), 1.001–0.965 (2H, m), 0.878–0.837 (3H, t, J = 6.2 Hz, terminal CH3); IR mmax/cm−1: 3330, 2900, 1700, 1420, 1160, 920, 740; [a]20 D −86.7 (c 2.05 mg ml−1 in CHCl3); MS m/z: 400 (M+, 49%), 385 (12), 329 (18), 315 (100), 175 (17); HR-MSm/z calculated forC25H36O4: 400.2614, found 400.2593.
(+)-CBD and its derivatives
(+)-CBD (4c). Basic aluminium oxide (15.6 g) was added to dry dichloromethane (150 ml). To this suspension BF3°§OEt2 (2.3 ml) was added under nitrogen. The mixture was stirred for 15 min at room temperature and then boiled for 1 min. To the boiling solution was added p-mentha-1,8-diene-3-ol (isopiperitenol) (950 mg, 6.25 mmol) and olivetol (1.35 g, 7.5 mmol) in dichloromethane (50 ml) and the reaction mixture was quenched within 10 sec with 10% aqueous solution of sodium bicarbonate (50ml). The organic part was separated and the aqueous layer was further extracted with dichloromethane. The combined dichloromethane solution was extracted with water, brine, dried (Na2SO4) and evaporated to give an oil. This oil was purified by silica gel column chromatography, using petroleum ether and ether as an eluant. Yield: 863 mg (44%); 1H-NMR (CDCl3, 300 MHz): d 0.90 (t, J = 7.5 Hz, 3H), 1.219– 1.329 (m, 7H), 1.529–1.603 (m, 2H), 1.660 (s, 3H), 1.794 (s, 3H), 2.00–2.210 (br t, 2H), 2.397–2.458 (m, 3H), 3.900 (br s, 1H), 4.556 (s, 1H), 4.658 (s, 1H), 4.90–5.00 (br, 1H, OH), 5.574 (s, 1H), 5.950–6.050 (br, 1H, OH), 6.10–6.30 (br, 2H, ArH); IR mmax/cm−1: 3425, 3000, 2930, 1630, 1145, 1380, 1219, 1025, 883; [a]20 D + 90 (c 3 mg ml−1 in EtOH); MS m/z: 314 (M+, 5%), 246 (13), 231 (100), 193 (9), 174 (9), 121 (10);HR-MSm/z calculated for C21H30O2: 314.2246, found 314.2212.
(+)-CBD-DMH (4d). 4d was prepared by the same procedure
as reported above for 4c, using DMH, instead of olivetol, as starting material. Yield: 55%; 1H-NMR (CDCl3, 300 MHz): d 0.832 (t, J = 7.5 Hz, 3H), 0.950–1.050 (br, 2H), 1.208 (br s, 12H), 1.454–1.505 (m, 2H), 1.635 (s, 3H), 1.794 (s, 3H), 2.050– 2.300 (m, 2H), 3.850 (br, 1H), 4.556 (s, 1H), 4.545 (s, 1H), 4.656 (s, 1H), 5.560 (s, 1H), 5.90–6.050 (br, 1H, OH), 6.250–6.358 (br, 2H, ArH); IR mmax/cm−1: 3450, 1680, 1580, 1445, 1380, 1219, 1025, 883; [a]20 D + 62 (c 8.1 mg ml−1 in MeOH); MS m/z: 370 (M+, 5%), 302 (13), 287 (100), 249 (18), 217 (25), 202 (14), 187 (10); HR-MS m/z calculated for C25H38O2: 370.2872, found 370.2832. Derivatives of (+)-CBD were prepared by the same procedure as reported for the (−)-CBD derivatives with the following yields: 5c (95%), 5d (96%), 6c (66%), 6d (70%), 7c (96%), 7d (97%), 8c (∼100%), 8d (∼100%), 9c (90%), 9d (90%), 10c (94%), 10d (92%), 11c (94%), 11d (94%), 12c (42%), 12d (42%), 13c (42%), 13d (58%), 14c (∼ 100%), 14d (∼ 100%), 15c (90%), 15d (90%), 16c (32%), 16d (30%), 17c (85%), 17d (86%), 18c (85%) and 18d (92%).
Acknowledgements
This work was supported by the US National Institute of Drug Abuse and the Israel Science Foundation.
View Larger
Table 1 Binding of (−)- and (+)-cannabidiol derivatives to the central (CB1) and peripheral (CB2) cannabinoid receptors

View Larger
Scheme 1
View Larger
Scheme 2 Reagents and conditions: (a) CH3I, K2CO3 in DMF; (b) 3-chloroperbenzoic acid in CH2Cl2; (c) methylmagnesium-N-cyclohexylisopropylamide in toluene; (d) Ac2O in pyridine; (e) TMSBr in CH2Cl2; (f) (nBu)4NH4OAc in acetone; (g) NaOH aq; (h) CH3MgI at 200 ◦C.
View Larger
Scheme 3 Reagents and conditions: (a) CH3MgI at 210 ◦C; (b) Ac2O in pyridine; (c) TMSBr in CH2Cl2; (d) K2CrO4 in HMPA; (e) NaClO2;(f) NaBH4 reflux in ethanol.
